СВОЙСТВА И СТРОЕНИЕ АЦЕТИЛЕНОВЫХ СОЕДИНЕНИЙ
Высокая эндотермичность реакции образования ацетилена из элементов (ΔH°298 = 226,7 кДж/моль)
2C(тв) + H2(г) = C2H2(г) - 226,7 кДж/моль (4)
делает ацетилен одним из самых богатых энергией углеводородов. Реакция образования этилена из элементов значительно менее эндотермична (ΔH°298 = 52 кДж/моль). Соответственно велика вероятность инициированного цепного разложения ацетилена на элементы по реакции, обратной (4), с выделением тепла, которое и лежит в основе взрыва, детонации и самовоспламенения ацетилена. Количество тепла, выделяемого при разложении 1 кг ацетилена, примерно в 1,5 раза больше, чем при разложении 1 кг нитроглицерина, и вдвое больше, чем для такого же количества тротила. Существующие нормы и технологические приемы позволяют тем не менее создавать взрывобезопасные процессы переработки ацетилена [8]. Например, в случае 100% ацетилена при давлении ниже 0,65 Па взрыв не вызывается при любых энергиях инициирования взрыва.
Высокая эндотермичность ацетилена приводит к высокой экзотермичности всех реакций присоединения по тройной связи. Энергия разрыва C≡C- и ≡C–H-связей составляют 961,4 и ~543 кДж/моль соответственно. При этом связь ≡C–H- является самой прочной среди любых C–H-связей в органических молекулах.
Спектры ядерного магнитного резонанса (на ядрах 1H и 13C), характеризующие степень экранирования этих ядер электронами, свидетельствуют о сильном смещении электронной плотности атома H к атому углерода. Об этом же говорят сравнение дипольных моментов связей ≡C–H (1D) и =C–H (0,6D) и результаты квантово-химического расчета эффективных (частичных) зарядов в молекулах C2H2 и C2H4:
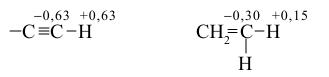
Большую полярность ацетиленовой C–H-связи объясняют более заметным вкладом s-орбиталей в гибридную орбиталь атома углерода в ацетилене (sp-гибридизация), чем в этилене (sp2-гибридизация). Известно, что s-орбиталь является более электроотрицательной, чем p-орбиталь (выше сродство этой орбитали к электрону). Приведенные выше особенности строения алкинов проявляются в такой характерной черте ацетилена и монозамещенных алкинов, как повышенная по сравнению с другими углеводородами кислотность (значение pKa алкинов находится в интервале 21-28):

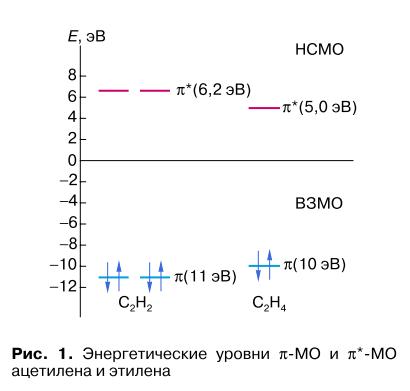
На рис. 1 представлена энергетическая диаграмма граничных мелекулярных орбиталей молекул ацетилена и этилена (о методе молекулярных орбиталей (МО) см. [9]). Молекула ацетилена содержит две одинаковые по энергии (вырожденные) высшие занятые π-МО (ВЗМО) и две низшие свободные МО (НСМО) разрыхляющего типа. Потенциал ионизации ацетилена (переход к катион-радикалу C2H2+•) составляет 11,4 эВ и выше, чем у этилена, на 1 эВ. Сродство к электрону ацетилена (переход к анион-радикалу C2H2-•) ниже, чем этилена, то есть ацетилен является более слабым донором и более слабым акцептором электрона, чем этилен.
Ацетилен реагирует с электроноакцепторными реагентами (электрофилами, E), электронодонорными реагентами - ионами и молекулами (нуклеофилами, Nu), с радикалами (H•, CH3•), атомами и комплексами металлов.
делает ацетилен одним из самых богатых энергией углеводородов. Реакция образования этилена из элементов значительно менее эндотермична (ΔH°298 = 52 кДж/моль). Соответственно велика вероятность инициированного цепного разложения ацетилена на элементы по реакции, обратной (4), с выделением тепла, которое и лежит в основе взрыва, детонации и самовоспламенения ацетилена. Количество тепла, выделяемого при разложении 1 кг ацетилена, примерно в 1,5 раза больше, чем при разложении 1 кг нитроглицерина, и вдвое больше, чем для такого же количества тротила. Существующие нормы и технологические приемы позволяют тем не менее создавать взрывобезопасные процессы переработки ацетилена [8]. Например, в случае 100% ацетилена при давлении ниже 0,65 Па взрыв не вызывается при любых энергиях инициирования взрыва.
Высокая эндотермичность ацетилена приводит к высокой экзотермичности всех реакций присоединения по тройной связи. Энергия разрыва C≡C- и ≡C–H-связей составляют 961,4 и ~543 кДж/моль соответственно. При этом связь ≡C–H- является самой прочной среди любых C–H-связей в органических молекулах.
Спектры ядерного магнитного резонанса (на ядрах 1H и 13C), характеризующие степень экранирования этих ядер электронами, свидетельствуют о сильном смещении электронной плотности атома H к атому углерода. Об этом же говорят сравнение дипольных моментов связей ≡C–H (1D) и =C–H (0,6D) и результаты квантово-химического расчета эффективных (частичных) зарядов в молекулах C2H2 и C2H4:
Большую полярность ацетиленовой C–H-связи объясняют более заметным вкладом s-орбиталей в гибридную орбиталь атома углерода в ацетилене (sp-гибридизация), чем в этилене (sp2-гибридизация). Известно, что s-орбиталь является более электроотрицательной, чем p-орбиталь (выше сродство этой орбитали к электрону). Приведенные выше особенности строения алкинов проявляются в такой характерной черте ацетилена и монозамещенных алкинов, как повышенная по сравнению с другими углеводородами кислотность (значение pKa алкинов находится в интервале 21-28):
На рис. 1 представлена энергетическая диаграмма граничных мелекулярных орбиталей молекул ацетилена и этилена (о методе молекулярных орбиталей (МО) см. [9]). Молекула ацетилена содержит две одинаковые по энергии (вырожденные) высшие занятые π-МО (ВЗМО) и две низшие свободные МО (НСМО) разрыхляющего типа. Потенциал ионизации ацетилена (переход к катион-радикалу C2H2+•) составляет 11,4 эВ и выше, чем у этилена, на 1 эВ. Сродство к электрону ацетилена (переход к анион-радикалу C2H2-•) ниже, чем этилена, то есть ацетилен является более слабым донором и более слабым акцептором электрона, чем этилен.
Ацетилен реагирует с электроноакцепторными реагентами (электрофилами, E), электронодонорными реагентами - ионами и молекулами (нуклеофилами, Nu), с радикалами (H•, CH3•), атомами и комплексами металлов.
РЕАКЦИИ АЛКИНОВ С ЭЛЕКТРОФИЛАМИ, НУКЛЕОФИЛАМИ И РАДИКАЛАМИ
Простейший электрофил H+ присоединяется в газовой фазе к алкинам с выделением тепла и образованием винильного карбокатиона (RC+=CH2) практически с нулевой энергией активации. В связи с этим большая реакционная способность алкенов по сравнению с ацетиленами по отношению к H+ (и другим электрофилам) объясняется термодинамическими причинами: более высоким положением ВЗМО в алкене (алкен - лучший донор электронов) и соответственно по сравнению с ацетиленами:
C2H2 + H+ → C2H3+ + 631,2 кДж/моль,
C2H4 + H+ → C2H5+ + 668,8 кДж/моль.
На первой стадии взаимодействия E+ (H+ или другого электрофила, например иона HgX+) с ацетиленом (схема 1) образуется симметричный π-комплекс, который:
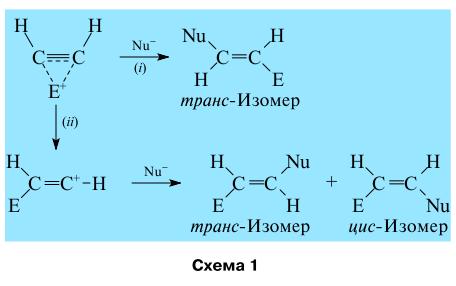
(i) - взаимодействует с Nu- с образованием продукта транс-присоединения частиц E+ и Nu- или (ii) - изомеризуется в классический ион карбения, присоединяющий Nu- с образованием смеси транс- и цис-изомеров. В π-комплексе H+ с алкинами наблюдается очень слабое цис-искажение алкина и очень незначительное изменение углов C–C–H и длин связей C–C.
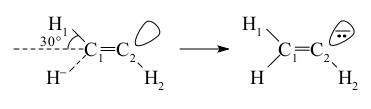
В отличие от реакции с электрофилами нуклеофильные реагенты быстрее присоединяются к ацетилену (алкинам), чем к этилену (алкенам). Этот экспериментальный факт, давно отмеченный химиками, явно противоречит распределениям зарядов, положению НСМО и значениям сродства к электрону в случае этих молекул. Загадка разрешилась очень просто (Р. Хоук, 1979 год). Обсуждая реакционную способность ацетилена в реакции присоединения к нему Nu, не следует рассматривать характеристики молекулы ацетилена в исходном состоянии, так как присоединение частицы с парой электронов (Nu) к богатой электронами тройной связи из-за сильного отталкивания электронов не может происходить без энергии активации и должно вызывать сильную перестройку молекулы алкина. Действительно, оказалось, что по мере приближения к тройной связи самого простого нуклеофила H- молекула ацетилена сильно изгибается и приобретает транс-конфигурацию:
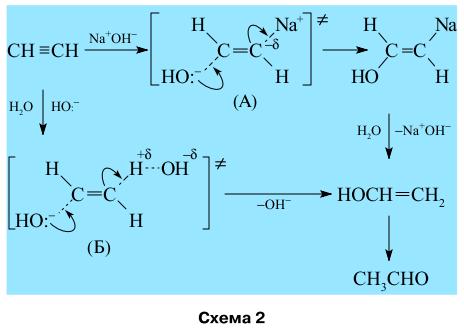
энергия НСМО снижается, заряд с атакуемого атома C1 вытесняется на C2 и H2, причем на молекулу ацетилена переносится значительная часть электронной плотности с нуклеофила (с H-). Таким образом, молекула C2H2 (слабый электрофил в исходном состоянии) становится сильным электрофилом в процессе взаимодействия с Nu вдоль пути реакции, причем более сильныи электрофилом, чем этилен, который незначительно меняет свою геометрию до барьера. Может иметь место также и сильное цис-искажение ацетилена в случае тех же нуклеофилов, но с существенно большей величиной энергетического барьера, и потому оно менее вероятно. Образующийся в результате присоединения нуклеофила винильный карбоанион уже в переходном состоянии может стабилизироваться противоионом (катионом) или электрофильной частью молекулы Nu–E. Например, ацетилен присоединяет молекулу воды в присутствии катализатора водного раствора NaOH (15 атм, 150°C) с участием двух вероятных переходных состояний (или активированных комплексов) (А) и (Б) (схема 2).
Присоединение электрона к молекуле C2H2 и радикалов (H•, CH3•) также происходит с транс-изгибом молекулы алкина, при этом радикальные частицы присоединяются к алкенам с несколько меньшей энергией активации, чем нуклеофилы.
На первой стадии взаимодействия E+ (H+ или другого электрофила, например иона HgX+) с ацетиленом (схема 1) образуется симметричный π-комплекс, который:
(i) - взаимодействует с Nu- с образованием продукта транс-присоединения частиц E+ и Nu- или (ii) - изомеризуется в классический ион карбения, присоединяющий Nu- с образованием смеси транс- и цис-изомеров. В π-комплексе H+ с алкинами наблюдается очень слабое цис-искажение алкина и очень незначительное изменение углов C–C–H и длин связей C–C.
В отличие от реакции с электрофилами нуклеофильные реагенты быстрее присоединяются к ацетилену (алкинам), чем к этилену (алкенам). Этот экспериментальный факт, давно отмеченный химиками, явно противоречит распределениям зарядов, положению НСМО и значениям сродства к электрону в случае этих молекул. Загадка разрешилась очень просто (Р. Хоук, 1979 год). Обсуждая реакционную способность ацетилена в реакции присоединения к нему Nu, не следует рассматривать характеристики молекулы ацетилена в исходном состоянии, так как присоединение частицы с парой электронов (Nu) к богатой электронами тройной связи из-за сильного отталкивания электронов не может происходить без энергии активации и должно вызывать сильную перестройку молекулы алкина. Действительно, оказалось, что по мере приближения к тройной связи самого простого нуклеофила H- молекула ацетилена сильно изгибается и приобретает транс-конфигурацию:
энергия НСМО снижается, заряд с атакуемого атома C1 вытесняется на C2 и H2, причем на молекулу ацетилена переносится значительная часть электронной плотности с нуклеофила (с H-). Таким образом, молекула C2H2 (слабый электрофил в исходном состоянии) становится сильным электрофилом в процессе взаимодействия с Nu вдоль пути реакции, причем более сильныи электрофилом, чем этилен, который незначительно меняет свою геометрию до барьера. Может иметь место также и сильное цис-искажение ацетилена в случае тех же нуклеофилов, но с существенно большей величиной энергетического барьера, и потому оно менее вероятно. Образующийся в результате присоединения нуклеофила винильный карбоанион уже в переходном состоянии может стабилизироваться противоионом (катионом) или электрофильной частью молекулы Nu–E. Например, ацетилен присоединяет молекулу воды в присутствии катализатора водного раствора NaOH (15 атм, 150°C) с участием двух вероятных переходных состояний (или активированных комплексов) (А) и (Б) (схема 2).
Присоединение электрона к молекуле C2H2 и радикалов (H•, CH3•) также происходит с транс-изгибом молекулы алкина, при этом радикальные частицы присоединяются к алкенам с несколько меньшей энергией активации, чем нуклеофилы.
РЕАКЦИИ АЛКИНОВ С КОМПЛЕКСАМИ ПЕРЕХОДНЫХ МЕТАЛЛОВ
Взаимодействуя с комплексами переходных металлов и ртути (II), ацетилены образуют огромное семейство π-комплексов, играющих важную роль в химии металлоорганических соединений и катализе [8] (см. также [10]). Количество молекул алкина (A) и атомов металла (или металлсодержащих фрагментов [M] меняется в π-комплексах в весьма широких пределах: [M]m(A)n, где m = 1-7, n = 1-4. Ацетиленовый лиганд во всех комплексах имеет цис-конфигурацию. Строение комплекса [M]–(A) (m = 1, n = 1) может быть представлено структурой (I), где [M]–MLq (L - неацетиленовые лиганды):
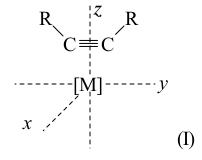
Длина связи C–C в координированном алкине в комплексах [M](A)n увеличивается (lC–C = 0,124-0,135 нм), а угол δ (∠C–C–R) изменяется в интервале 128-165°. Напомним, что длина связи C–C в олефине (алкене) составляет ~0,134 нм, а угол δ равен ~120°. Таким образом, воздействие металла на алкин приводит к тому, что заметно меняется гибридизация электронов у атома углерода и алкин (sp-гибридизация) по своей геометрии приближается к алкену (sp2-гибридизация). Цис-искажение алкина в π-комплексе не только делает более удобной координацию алкина металлом, поскольку заместители при тройной связи не мешают физически лигандам, связанным с металлом (MLq), но и приводит к повышению донорных и акцепторных свойств алкина. При цис-искажении геометрии алкина в π-комплексе (так же как и в циклоалкинах: циклогептине, циклооктине) снимается вырождение π- и π*-МО ив результате повышается энергия ВЗМО и снижается энергия НСМО (рис. 2). Таким образом, алкин в π-комплексе может быть донором двух пар π-электронов (π⊥ и π‖) на подходящие по симметрии орбитали металла с образованием двух донорно-акцепторных связей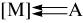 и акцептором двух пар d-электронов с подходящих по симметрии d-орбиталей металла с образованием двух так называемых дативных связей
и акцептором двух пар d-электронов с подходящих по симметрии d-орбиталей металла с образованием двух так называемых дативных связей  . Заштрихованные и незаштрихованные части граничных поверхностей орбиталей, из которых посторены МО цис-алкина на рис. 2, обозначают различные знаки (±) угловой части волновой функции электронов в различных областях пространства. Напомним, что в атомах d-элементов (IV-VI периоды Периодической системы элементов Sc→Zn, Y→Cd и Lu→Hg) имеются пять d-орбиталей, на которых может быть расположено максимально 10 d-электронов (d10). Граничные поверхности d-орбиталей, то есть поверхности, охватывающие области наибольшей электронной плотности, приведены на рис. 3 (координаты x, y, z структуры I). Комплексы металлов начала переходных серий, имеющие d2-d6 электронов, не участвуют в образовании связей с другими (кроме А)
. Заштрихованные и незаштрихованные части граничных поверхностей орбиталей, из которых посторены МО цис-алкина на рис. 2, обозначают различные знаки (±) угловой части волновой функции электронов в различных областях пространства. Напомним, что в атомах d-элементов (IV-VI периоды Периодической системы элементов Sc→Zn, Y→Cd и Lu→Hg) имеются пять d-орбиталей, на которых может быть расположено максимально 10 d-электронов (d10). Граничные поверхности d-орбиталей, то есть поверхности, охватывающие области наибольшей электронной плотности, приведены на рис. 3 (координаты x, y, z структуры I). Комплексы металлов начала переходных серий, имеющие d2-d6 электронов, не участвуют в образовании связей с другими (кроме А)
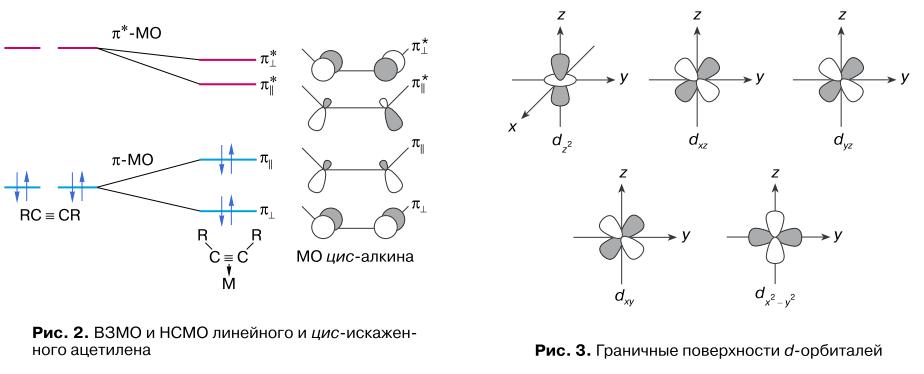
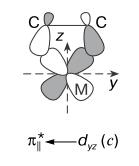
лигандами (несвязывающая d2-d6-конфигурация металла), образуют наиболее устойчивые π-комплексы за счет связей π⊥ → dxz (a), π‖ → dz2 (b) и π‖* ← dyz (c) (см. рис. 2 и 3). Например, образование связи с-типа (перекрывание занятой d-электронами dyz-орбитали металла с разрыхляющей вакантной π‖*-МО цис-алкина) представлено диаграммой:
Комплексы металлов конца переходных серий образуются за счет связей b- и c-типа, причем вакантной орбиталью металла могут быть (вместо dz2) dsp-, dp- и s-орбитали. При этом в π-комплексах Ni(II), Pd(II), Pt(II), Cu(I), Ag(I), Au(I) и, вероятно, Hg(II) основной вклад в энергию связи вносит перенос M ← A (b-тип), а в π-комплексах Fe(0,II), Co(0,I), Ni(0), Rh(I), Pd(0), Os(0,I), Ir(I), Pt(0) основной вклад определяется переносом M → A (c-тип). Преимущественное направление переноса заряда определяет поляризацию координированного ацетилена. Так, в случае b-типа ацетилен становится лучшим электрофилом, его H-атомы становятся более кислыми. В случае c-типа (дативная связь) на ацетилене появляется избыточная электронна плотность - в пределе образуется металлоцикл (металлациклопропен) с двумя σ-связями МС (структура (II) (анион R2C22- и катион L2Ni2+):
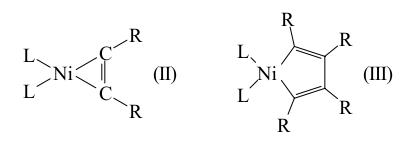
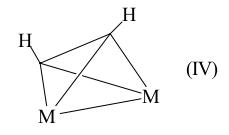
Естественно, такой алкин будет легче реагировать с электрофильными частицами (H+) или другими ненасыщеными молекулами (например, алкинами) с образованием структуры (III) как типичный металлоорганический реагент, содержащий группу M–R и способный внедрять алкин по связи M–C. Чем больше атомов металла в π-комплексе со связями M–M (кластеры, см. [11]), тем сильнее воздействие на ацетилен, тем сильнее меняются его геометрия, энергетические характеристики и реакционная способность. В таких комплексах алкин является мостиком между 2, 3 и 4 атомами металла (обозначается приставкой μ2-, μ3- и μ4-). Угол C–C–R достигает 114°, а длина C≡C-связи приближается к длине простой связи C–C (0,145 нм). π-Комплексы состава M2(μ2–А) могут в пределе иметь структуру диметаллатетраэдрана (IV), например W2(OR)6Py2(C2H2) (lC-C = 0,141 нм):
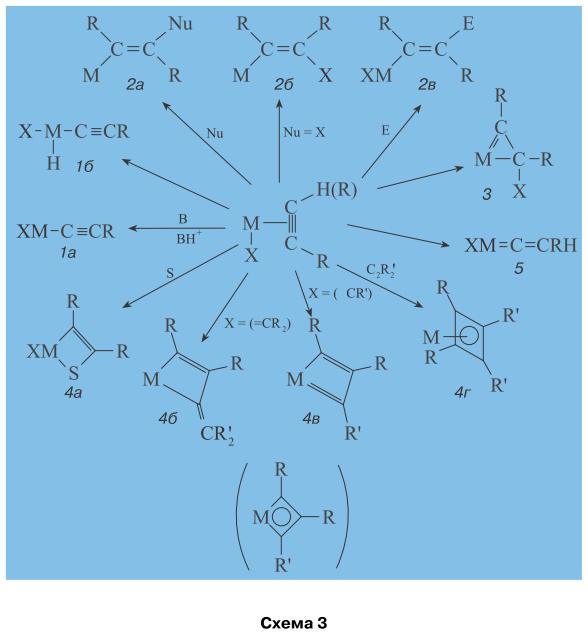
Образование дативных связей (M → A) для активации алкина особенно важно, поскольку перенос M → A не только упрочняет сам π-комплекс и меняет поляризацию алкина,но, кроме того, сильно дестабилизирует, активирует алкин, вовлекая во взаимодействие разрыхляющую π*-МО алкина [3]. Известние пути превращения ацетиленовых π-комплексов, в которых и происходит активация C≡C- и ≡C–H-связей, и строение образующихся металлоорганических соединений приведены на схеме 3. Направление реакций определяется природой металл, его мтепенью окисления, природой лигандов, группы X и других реагентов (E, Nu, S, B).
Среди продуктов превращения первичных π-комплексов мы видим соединения, образующиеся при транс-присоединении Nu (2а), E (2в) и цис-присоединении нуклеофильного реагента X, связанного с металлом (2б). В ряде случаев σ-винильное производное металла (2) превращается в металлациклопропеновый продукт (3), содержащий карбеновый (винилидиеновый) лиганд. Образование этинильных соединений происходит в результате кислотной диссоциации ≡C–H-связи и замещения иона водорода ионом металла (если металл является акцептором электронов и повышает положительный эффективный заряд на H-атоме) (1а) или в результате окислительного присоединения алкина к комплексу металла с разрывом ≡C–H-связи (1б). Изомеризация ацетилена в винилиден (CH2=C:) - процесс эндотермический. Энергия перехода в синглетный винилиден (все электроны спарены) (реакция (5)) составляет ~175 кДж/моль,
HC≡CH → CH2=C: - 175 кДж/моль, (5)
поэтому в свободном состоянии такие молекулы практически не существуют. Однако в координационной сфере π-комплекса происходит стабилизация винилиденовой группы, и такие соединения (5) легко образуются и участвуют в каталитических реакциях. Образование металлоциклов (на примере моноядерных по металлу соединений) представлено реакциями внедрения молекул S (алкин, алкен, CO2, RNCO, CO, RNC и др.) по связи M–C π-комплексов со структурой II (4а) и присоединения алкинов к карбеновым (4б) или карбиновым (4в) лигандам X. Характерной реакцией для π-комплексов Pd(II), Ni(II), Fe(0) и других металлов является образование π-циклобутадиеновых комплексов (4г). Подробнее механизмы образования и превращений этих соединений описаны в работе [8]. π-Комплексы и σ-металлоорганические соединения алкинов возникают и при взаимодействии алкинов с поверхностью металлов (металлических катализаторов) [8, 12]. Физическими методами обнаружены π-комплексы с одним, тремя и четырьмя атомами металла на поверхности, этинильные (RC≡C–M) и винилиденовые (CH2=C=M) фрагменты. Все образующиеся из π-комплексов соединения встречаются в качестве промежуточных продуктов в металлоорганических и каталитических синтезах с участием алкинов.
Длина связи C–C в координированном алкине в комплексах [M](A)n увеличивается (lC–C = 0,124-0,135 нм), а угол δ (∠C–C–R) изменяется в интервале 128-165°. Напомним, что длина связи C–C в олефине (алкене) составляет ~0,134 нм, а угол δ равен ~120°. Таким образом, воздействие металла на алкин приводит к тому, что заметно меняется гибридизация электронов у атома углерода и алкин (sp-гибридизация) по своей геометрии приближается к алкену (sp2-гибридизация). Цис-искажение алкина в π-комплексе не только делает более удобной координацию алкина металлом, поскольку заместители при тройной связи не мешают физически лигандам, связанным с металлом (MLq), но и приводит к повышению донорных и акцепторных свойств алкина. При цис-искажении геометрии алкина в π-комплексе (так же как и в циклоалкинах: циклогептине, циклооктине) снимается вырождение π- и π*-МО ив результате повышается энергия ВЗМО и снижается энергия НСМО (рис. 2). Таким образом, алкин в π-комплексе может быть донором двух пар π-электронов (π⊥ и π‖) на подходящие по симметрии орбитали металла с образованием двух донорно-акцепторных связей
лигандами (несвязывающая d2-d6-конфигурация металла), образуют наиболее устойчивые π-комплексы за счет связей π⊥ → dxz (a), π‖ → dz2 (b) и π‖* ← dyz (c) (см. рис. 2 и 3). Например, образование связи с-типа (перекрывание занятой d-электронами dyz-орбитали металла с разрыхляющей вакантной π‖*-МО цис-алкина) представлено диаграммой:
Комплексы металлов конца переходных серий образуются за счет связей b- и c-типа, причем вакантной орбиталью металла могут быть (вместо dz2) dsp-, dp- и s-орбитали. При этом в π-комплексах Ni(II), Pd(II), Pt(II), Cu(I), Ag(I), Au(I) и, вероятно, Hg(II) основной вклад в энергию связи вносит перенос M ← A (b-тип), а в π-комплексах Fe(0,II), Co(0,I), Ni(0), Rh(I), Pd(0), Os(0,I), Ir(I), Pt(0) основной вклад определяется переносом M → A (c-тип). Преимущественное направление переноса заряда определяет поляризацию координированного ацетилена. Так, в случае b-типа ацетилен становится лучшим электрофилом, его H-атомы становятся более кислыми. В случае c-типа (дативная связь) на ацетилене появляется избыточная электронна плотность - в пределе образуется металлоцикл (металлациклопропен) с двумя σ-связями МС (структура (II) (анион R2C22- и катион L2Ni2+):
Естественно, такой алкин будет легче реагировать с электрофильными частицами (H+) или другими ненасыщеными молекулами (например, алкинами) с образованием структуры (III) как типичный металлоорганический реагент, содержащий группу M–R и способный внедрять алкин по связи M–C. Чем больше атомов металла в π-комплексе со связями M–M (кластеры, см. [11]), тем сильнее воздействие на ацетилен, тем сильнее меняются его геометрия, энергетические характеристики и реакционная способность. В таких комплексах алкин является мостиком между 2, 3 и 4 атомами металла (обозначается приставкой μ2-, μ3- и μ4-). Угол C–C–R достигает 114°, а длина C≡C-связи приближается к длине простой связи C–C (0,145 нм). π-Комплексы состава M2(μ2–А) могут в пределе иметь структуру диметаллатетраэдрана (IV), например W2(OR)6Py2(C2H2) (lC-C = 0,141 нм):
Образование дативных связей (M → A) для активации алкина особенно важно, поскольку перенос M → A не только упрочняет сам π-комплекс и меняет поляризацию алкина,но, кроме того, сильно дестабилизирует, активирует алкин, вовлекая во взаимодействие разрыхляющую π*-МО алкина [3]. Известние пути превращения ацетиленовых π-комплексов, в которых и происходит активация C≡C- и ≡C–H-связей, и строение образующихся металлоорганических соединений приведены на схеме 3. Направление реакций определяется природой металл, его мтепенью окисления, природой лигандов, группы X и других реагентов (E, Nu, S, B).
Среди продуктов превращения первичных π-комплексов мы видим соединения, образующиеся при транс-присоединении Nu (2а), E (2в) и цис-присоединении нуклеофильного реагента X, связанного с металлом (2б). В ряде случаев σ-винильное производное металла (2) превращается в металлациклопропеновый продукт (3), содержащий карбеновый (винилидиеновый) лиганд. Образование этинильных соединений происходит в результате кислотной диссоциации ≡C–H-связи и замещения иона водорода ионом металла (если металл является акцептором электронов и повышает положительный эффективный заряд на H-атоме) (1а) или в результате окислительного присоединения алкина к комплексу металла с разрывом ≡C–H-связи (1б). Изомеризация ацетилена в винилиден (CH2=C:) - процесс эндотермический. Энергия перехода в синглетный винилиден (все электроны спарены) (реакция (5)) составляет ~175 кДж/моль,
поэтому в свободном состоянии такие молекулы практически не существуют. Однако в координационной сфере π-комплекса происходит стабилизация винилиденовой группы, и такие соединения (5) легко образуются и участвуют в каталитических реакциях. Образование металлоциклов (на примере моноядерных по металлу соединений) представлено реакциями внедрения молекул S (алкин, алкен, CO2, RNCO, CO, RNC и др.) по связи M–C π-комплексов со структурой II (4а) и присоединения алкинов к карбеновым (4б) или карбиновым (4в) лигандам X. Характерной реакцией для π-комплексов Pd(II), Ni(II), Fe(0) и других металлов является образование π-циклобутадиеновых комплексов (4г). Подробнее механизмы образования и превращений этих соединений описаны в работе [8]. π-Комплексы и σ-металлоорганические соединения алкинов возникают и при взаимодействии алкинов с поверхностью металлов (металлических катализаторов) [8, 12]. Физическими методами обнаружены π-комплексы с одним, тремя и четырьмя атомами металла на поверхности, этинильные (RC≡C–M) и винилиденовые (CH2=C=M) фрагменты. Все образующиеся из π-комплексов соединения встречаются в качестве промежуточных продуктов в металлоорганических и каталитических синтезах с участием алкинов.
ЛИТЕРАТУРА
1. Кнунянц И.Л. Ацетилен // Хим. энциклопедия. М.: Сов. энциклопедия, 1998. Т.1.
2. Ньюленд Ю., Фогт Р. Химия ацетилена. М.: Изд-во иностр. лит., 1947. 400с.
3. Темкин О.Н., Флид Р.М. Каталитические превращения ацетиленовых соединений в растворах комплексов металлов. М.: Наука, 1968. 212 с.
4. Печуро Н.С., Песин О.Ю.// Итоги науки и техники. Технология орган. веществ. 1984. Т.9. С. 60-120.
5. Караханов Э.А.Что такое нефтехимия // Соросовский Образовательный Журнал. 1996. №2. С. 65-73.
6. Темкин О.Н. Промышленный катализ и экологически безопасные технологии // Там же. №10. С. 42-50.
7. Новые процессы органического синтеза / Под ред. С.П. Черных. М.: Химия, 1989. 399с.
8. Темкин О.Н., Шестаков Г.К., Трегер Ю.А. Ацетилен: Химия. Механизмы реакций. Технология. М.: Химия, 1991. 416с.
9. Витковская Н.М. Метод молекулярных орбиталей: Основные идеи и важные следствия // Соросовский Образовательный Журнал. 1996. №6. С. 58-64.
10. Штейнгарц В.Д. Координационный катализ в химии ненасыщенных соединений // Там же. №7. С. 47-58.
11. Темкин О.Н. Каталитическая химия // Там же. №1. С. 57-65.
12. Zaera F.// Chem. Rev. 1995. Vol. 95. P. 2651-2693.
2. Ньюленд Ю., Фогт Р. Химия ацетилена. М.: Изд-во иностр. лит., 1947. 400с.
3. Темкин О.Н., Флид Р.М. Каталитические превращения ацетиленовых соединений в растворах комплексов металлов. М.: Наука, 1968. 212 с.
4. Печуро Н.С., Песин О.Ю.// Итоги науки и техники. Технология орган. веществ. 1984. Т.9. С. 60-120.
5. Караханов Э.А.Что такое нефтехимия // Соросовский Образовательный Журнал. 1996. №2. С. 65-73.
6. Темкин О.Н. Промышленный катализ и экологически безопасные технологии // Там же. №10. С. 42-50.
7. Новые процессы органического синтеза / Под ред. С.П. Черных. М.: Химия, 1989. 399с.
8. Темкин О.Н., Шестаков Г.К., Трегер Ю.А. Ацетилен: Химия. Механизмы реакций. Технология. М.: Химия, 1991. 416с.
9. Витковская Н.М. Метод молекулярных орбиталей: Основные идеи и важные следствия // Соросовский Образовательный Журнал. 1996. №6. С. 58-64.
10. Штейнгарц В.Д. Координационный катализ в химии ненасыщенных соединений // Там же. №7. С. 47-58.
11. Темкин О.Н. Каталитическая химия // Там же. №1. С. 57-65.
12. Zaera F.// Chem. Rev. 1995. Vol. 95. P. 2651-2693.
* * *
Соросовский образовательный журнал