Химия и Химики № 1 2019
Журнал Химиков-Энтузиастов
| Оглавление | Видео опыты по химии | Видео опыты по физике | На главную страницу |
|
Химия и Химики № 1 2019 Журнал Химиков-Энтузиастов |
Химические истории: гальванические ванны и их анализ (аналитическая химия) ч.13-ч.16 Chemical stories: galvanic baths and their analysis (analytical chemistry) В.Н. Витер |
|
Обнаружив ошибку на странице, выделите ее и нажмите Ctrl + Enter
Твердое анодирование алюминия (анализ ванны)
Эту ванну технологи почему-то называют "твердое травление", почему так и для чего она - на момент написания статьи не разобрался (было не до этого), знал только, что состав ванны: вода и серная кислота (180-200 г/л). Находится в отделе серебрения. Судя по наличию электродов, ванна электрохимическая. Ни одного точного соответствия на термин "твердое травление" в гугле не нашел (ни на украинском, ни на русском), по-видимому, - местный жаргон. Хотя, все более склоняюсь к мнению, что я просто неправильно услышал название ванны.
Hard anodizing of aluminum (analysis of bath) Уже после написания статьи узнал правильное название данной ванны - "твердое анодирование алюминия". В результате сразу же удалось найти описание процесса. Твердое, или глубокое анодирование - электрохимическое формование на поверхности алюминия и его сплавов прочной оксидной пленки (толщиной 40-300 мкм). Пленка отличается твердостью, износостойкостью, жаростойкостью, хорошей коррозийной устойчивостью, но вместе с тем она хрупкая. Твердое анодирование обычно проводят в 17-30% растворах серной кислоты. У нас раньше в электролит добавляли также щавелевую кислоту, но потом от этой практики отошли. В процессе работы возникает проблема - сильное растравливание формирующейся оксидной пленки, скорость этого процесса возрастает при увеличении температуры. Поэтому электролит охлаждают до минусовых температур. Наиболее толстые, твердые оксидные пленки с наименьшей пористостью образуются при температуре растворах от -4 до -8°С. Необходимость охлаждения электролита создает для нашего предприятия проблему: заказы поступают редко, поэтому приходится содержать в рабочем состоянии холодильную установку, которая работает всего несколько дней в году. А когда работает - потребляет много энергии. И не только энергии. Недавно запуск ванны пришлось отложить из-за того, что на заводе не было фреона. Итак, химический анализ ванны. Методики в журнале нет, но, если там только серная кислота, и так все ясно: берешь аликвоту разведенного в 10 раз раствора и титруешь 0.1N раствором едкого натра. Можно - с метиловым оранжевым, но лучше - с фенолфталеином: сильную кислоту титруем щелочью - скачек рН в точке эквивалентности резкий и попадает в область перехода цвета обеих индикаторов. Зато с фенолфталеином заметить изменение цвета проще (бесцветный - малиновый против красный - оранжевый в случае метилоранжа). Даже коэффициент пересчета (миллилитров едкого натра в граммы на литр серной кислоты) рассчитывать не надо: можно взять с других аналогичных методик. Сходил с технологом, отобрал пробу раствора из ванны "твердого анодирования". По дороге заметил закрытую емкость с подписью "ванна амальгамирования". Считать, сколько примерно пойдет едкого натра на титрование, поленился, поэтому взял аликвоту в 1 мл неразбавленного раствора из ванны. На титрование аликвоты ушло 48.4 мл 0.1N раствора щелочи - это при том, что объем пипетки, которой я титрую - 10 мл. Набираешь - титруешь - опять набираешь и титруешь. Посчитал содержание серной кислоты - 239 г/л - ванну нужно разбавить. Сказал результат технологам и занялся другими делами.
Через неделю просят опять проанализировать. Странно, учитывая, что обычно эта ванна работает пару раз в год (да и то не каждый год). Спускаюсь - на дверях замок и опломбировано. Возвращаюсь к технологу: "Замок не захлопнут, пломбу - снимите, потом опять налепите". Отобрал ванну, пошел анализировать. В этот раз уже развел раствор в 10 раз (20 мл ванны в мерную колбу на 200 мл и водой до метки) - долго титровать не пришлось. Еще до расчетов вижу, что кислоты в ванне почти столько же, сколько и было (ну чуть меньше) - 233 г/л. Иду к технологам. - Вы что, проверить меня решили? (Даю бумажку с результатом). - Нет. - Ванну должны были разбавить? - Да. - Содержание кислоты 233 г/л - чуть меньше, но не на много (что разница почти в пределах точности анализа, выполненного в таких условиях, - молчу). - Я точно видела, как гальваник вылил в ванну ведро воды... - Одно на ванну? -Да. Мы сейчас разбавим, не могли бы вы сделать еще анализ? Другой технолог: - Нет времени. Будем работать, как есть: ванна уже как-то работала выше верхнего предела. Позже технолог призналась, что на анодной штанге вешают полоски, на них остаются "отметки", по которым можно определить, каким раньше был уровень раствора. Полоски, конечно, меняют, но редко. Рабочий как-раз и хотел добавить в ванну воды "до метки" (как сказали бы аналитики), но другой технолог ему помешала, т.к. боялась, что ванна выйдет разведенной. Гальваник успел влить только ведро воды.
В лаборатории есть даже СФ-46 - тоже старый, но не настолько древний фотометр (в нем даже дифракционная решетка есть), только для моих целей выделили именно КФК-2. |

Фотометр СФ-46 (действующий музейный экспонат) |
|
Узнай я марку прибора раньше, можно было бы скачать паспорт и посмотреть, но тогда я не знал - пришлось нервничать, угадывать и смотреть в интернете, где что. Спросить было у кого - в лаборатории есть еще зав. лаб. и химик, но этот вариант я оставил на крайний случай, т.к. обращаться к людям для меня очень трудно. Кроме того, в лаборатории был и остается конфликт интересов: заведующая и другой химик воспринимают лабораторию, как "комнату отдыха" (работа у них бумажная: обе по факту - технологи, химик, правда, изредка делает анализы, но не часто). Я же воспринимаю лабораторию, как "комнату для проведения анализов".
После предшественницы остался даже калибровочный график по палладию, только выглядит он немного странно: по оси абсцисс - миллиграммы палладия (концентрация), по оси ординат какая-то величина "С". - А там должна быть оптическая плотность раствора, которую обычно обозначают "D". Диапазон значений концентрации палладия ("миллиграммы" - ось абсцисс) почему-то не совпадал с тем, что было в методике из тетрадки моей предшественницы. Ни обозначения величин, ни их значения не совпадают - вот и думай, что это за график. Хочешь-не-хочешь, а пришлось спрашивать. Заведующая: "А кто его знает? Может она и не делала. Заставили - нарисовала сказочку". Химик принесла мне ОСТ с методикой анализа хлорида палладия. Она оказалась идентичной методике в тетрадке. - Не считая того, что в ОСТ используются 100-мл мерные колбы, а в тетрадке - 50 мл. |

Методика фотометрического определения палладия |

|
|
Ну и еще. У предшественницы диапазон концентраций палладия в ванне активации был 200-250 г/л. Сначала я не обратил внимание - много методик и я занимался другими. Но перед анализом понял: в тетрадке ошибка. Вместо "мг" написано "г": хорошо, что догадался. Сколько там должно быть палладия - я не знал, но если бы в ванне было 250г/л палладия, то возле нее стояло бы пару автоматчиков, а на 250 мг/л - мало кто позарится.
|

Методика фотометрического определения палладия (тетрадка предшественницы) |
|
Стандартный раствор палладия для построения калибровочного графика - опять пришлось просить: "PdCl2 1 г/л". Дали бутылку.
- А это "1 г/л" в пересчете на палладий или на хлорид? - Не помню. Посмотрите ОСТ - раствор готовили по ОСТу. Посмотрел - оказалось на хлорид. Отобрал 10 мл исходного раствора (т.е. 10 мг хлорида палладия), развел соляной кислотой в мерной колбе на 100 мл. 10 мл разведенного раствора - содержат 1 мг. По методике нужно взять от 0.1 до 0.7 мг хлорида палалдия, т.е. - 1-7 мл разведенного раствора. Чтобы построить калибровочный график на фотометрию, нужно 5-7 точек. А мерных колб на 50 мл - всего 3. Значит, будет всего 3 точки. Мерных колб на 100 мл сначала не нашел вообще (хотя бы 1 колба была нужна, чтобы разбавить исходный раствор хлорида палладия). К счастью, 100-мл колба потом нашлась - пришлось ее долго мыть... Выбрал точки: 2, 4 и 7 мл (0.2, 0.4 и 0.7 мг хлорида палладия). Перенес палладий пипеткой в колбы (горлышко оказалось насколько узкое, что только и смотри, чтобы раствор не пролился мимо). Добавил хлорид олова (II) и соляную кислоту (1:4). Добавил 3%-ю перекись водорода, которую приготовил из 60%-й. Нужно кипятить. - На водяной бане или ставить колбы на плитку? - Ставьте на плитку, только следите, чтобы не вскипели. Закипятил и прокипятил до разложения перекиси водорода, охлаждаю. Тем временем включил КФК-2. Выставил светофильтр 400 нм (синий). Наливаю в кювету воду, а стрелка не реагирует - она в зашкале. Стал сомневаться: на КФК-3 ноль выставляется нажатием одной кнопочки "Ноль", а как тут? Тут есть два рычага "Установить 100%". Оптическая плотность точно начинается с "0"? Посмотрел графики в интернете - да. Ставлю вместо кюветы непрозрачный предмет, потом убираю - стрелка не реагирует (она и так в зашкале). Пришлось спрашивать у зав. лаба. - Оптическая плотность же начинается с нуля, а тут - стрелка в противоположном конце. - А вы пробовали установить ноль ручкой регулирования? - Нет. Боялся сделать что-то не так. Пробует выставить ноль, но стрелка смешается максимум к средине шкалы - не дальше. - А прибор вообще рабочий? (Смотрю на цветочный горшок сверху и представляю, сколько воды могло попасть на фотометр, когда растение поливали...) - Должен работать. Меняет светофильтры - стрелка кое-как зашевелилась и позволила вывести себя на "0". Вернула синий светофильтр - стрелка резко отклонилась, но теперь ее удалось вернуть "0". Процедура заняла минут 15. Подождал полного охлаждения растворов, добавил в колбы йодид калия и аскорбиновую кислоту (раствор аскорбиновой кислоты готовят перед самым анализом - она легко окисляется). Растворы в колбе окрасились в красно-коричневый цвет. Довел до метки, перемешал. Померил оптическую плотность. Судя по значениям оптической плотности, точки вроде бы попадают на прямую, но нужно еще построить график и убедиться. |

Построение калибровочного графика - анализ палладия |

|

|

|

|

|

|

|

|

|

|

|

|

|

|

|

|

|

|

|

|

|

|

|

|

|
|
Построил калибровочный график - точки легли нормально. Сравнил с графиком предшественницы. После долгих раздумий понял, что ее график соответствует маленькому прямоугольнику в левой нижней части моего графика. Т.е., она его действительно построила, а не "нарисовала".
|
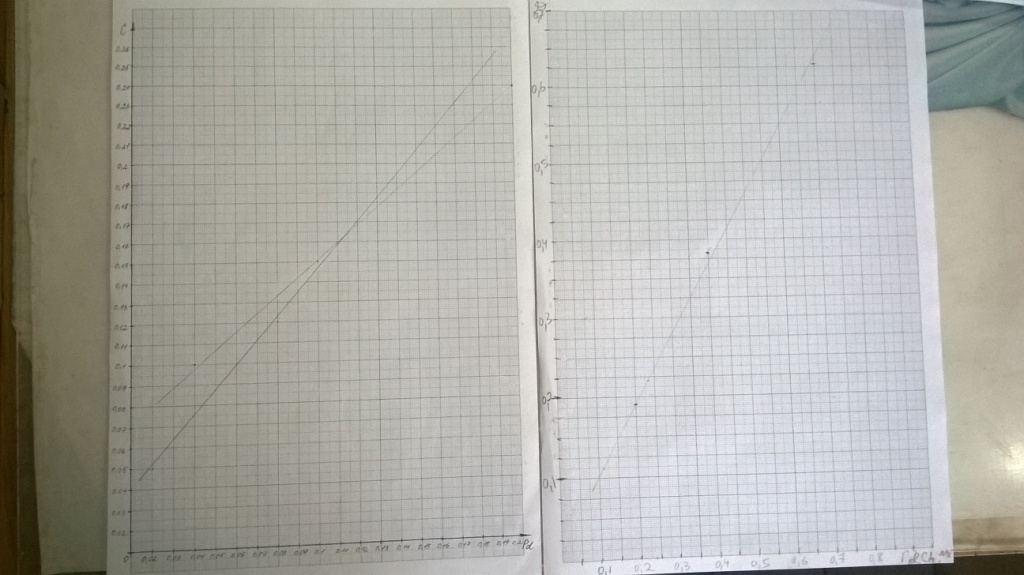
Калибровочный график для фотометрического анализа палладия (слева - моей предшественницы, справа - мой) |
|
График построил, но это только график. На следующий день - анализ ванны активации. Хорошо, что вовремя заметил: вместо граммов в составе ванны должны стоять миллиграммы - а то бы ждал меня очередной сюрприз. Там методика немного другая: берется 0.5, 1.0 и 1.5 мл раствора из ванны + 20 мл соляной кислоты (1:4), нагреть до кипения. Осторожно добавляем перекись водорода, кипятим до ее разложения. При добавлении перекиси водорода коричневый раствор бледнеет (там, где палладия меньше - обесцвечивается). Такое впечатление, что хлор (продукт реакции соляной кислоты и перекиси) реагирует с "палладиевыми кластерами" - как их назвали в технологическом регламенте (вероятно хлорид палладия с хлоридом двухвалентного олова дает коллоидный палладий). Потом - охлаждаем, добавляем йодид калия и аскорбиновую кислоту. Проводим фотометрию красно-коричневого раствора.
|

Фотометрический анализ палладия |

|

|

|

|

|

|

|

|

|

|

|

|

|

|

|

|

|

|

|
|
Как раз пришел начальник. Спрашивает результаты. Считаю. Взял график, смотрю, сколько хлорида палладия. Пересчитываю три параллельных на 1 мл раствора, потом пересчитываю с хлорида палладия на палладий (коэффициент 0.6). Еще в процессе расчетов вижу, что не выходит: был бы график не по хлориду, а по палладию - при таких цифрах было бы все нормально. Но график по хлориду и выходит, что палладия - 141 мг/л при норме 200-250.
Хорошо, что рядом был начальник. Говорю: - Не получается. Мало. - Сколько? - 141. - Так это нормально: линия работает и при более низком палладии. - Не было бы вас здесь, я бы нервничал и искал, где ошибка в анализе... - Так можно же позвонить и спросить. Можно. Можно, конечно, - если знать, что спрашивать. |
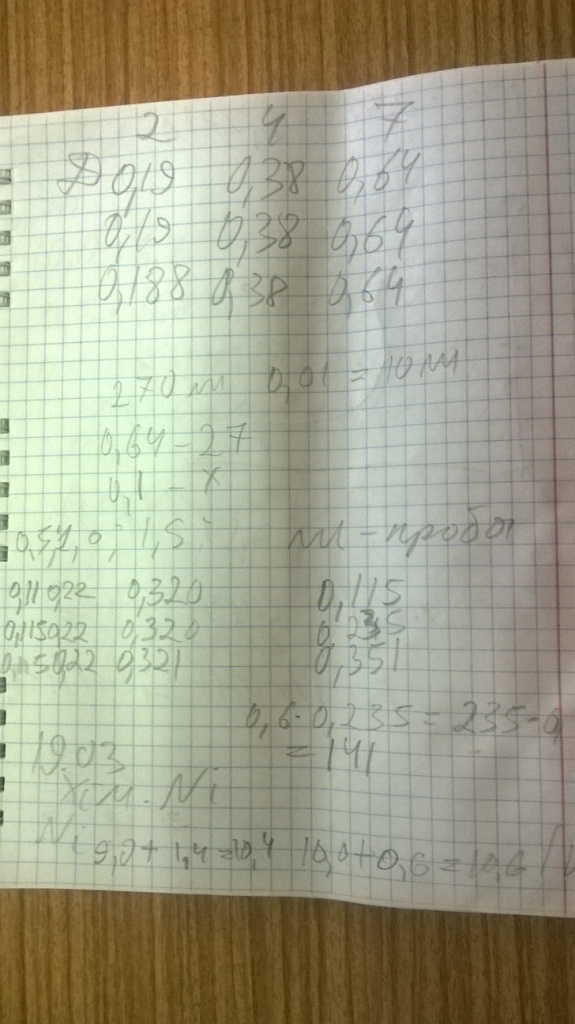
Мои расчеты |
|
Через пару недель снова делал анализ палладиевого активатора. В этот раз получилось 121 мг/л. Начальник сказал - пускай так и работает, сделаешь анализ через неделю. Но линия остановилась через несколько дней - "не тянется" медь. Я как раз только успел отобрать пробу из ванны хромирования (там возникли проблемы) и начать анализ, как звонит начальник - нужно проанализировать палладий и олово. Говорю: сначала я сделаю ЭТО (т.е. - хром), а потом уже - ванну активации. Начальник - хорошо, конечно. Сделал ванну хромирования, прибегает начальник. Палладий? - Начал делать.
Снова спешка. Отобрал в 3 колбы 0.5, 1.0 и 1.5 л раствора. Добавил соляной кислоты. В методике у предшественницы написано "конц. (1:4) HCl". Я прочитал наспех и взял просто концентрированную солянку. Нагрел до кипения, добавляю 60%-ю перекись. Чувствую, что-то сильный запах хлора - аж слишком. И раствор стал желтым. Понял ошибку - нужно было брать соляную кислоту (1:4), а не концентрированную. Сначала думал - хлор улетит, раствор побледнеет. Кипячу раствор, а он все остается желтым. Взял три другие колбочки (спасибо коллеге - они уже были) и сделал анализ заново. И опять не все прошло гладко. После кипячения (необходимого, чтобы разложилась перекись) охладил раствор и добавил йодид калия - смотрю, а в колбе, где аликвота анализируемого раствора была меньше (0.5 мл), раствор окрашен интенсивнее, чем в колбе, где аликвота была больше (1.0 мл). Добавляю аскорбиновую кислоту - раствор в первой колбе побледнел и стал "каким надо". В чем дело? И тогда я понял, зачем нужна аскорбиновая кислота. Если перекись водорода при кипячении разложилась не до конца (скорее, скорее - линия металлизации стоит!).., перекись водорода вытесняет йод из йодида калия - он и дает окраску. Аскорбиновая кислота снова восстанавливает йод до йодида. "Лишняя" окраска исчезает. Меряю оптическую плотность - а она даже чуть выше, чем было в прошлый раз. Следующие параллельные - чуть ниже. В среднем концентрация не поменялась (в пределах точности). Олово? Титрую олово - оно упало на 0.6 г/л. Значит, состав почти не поменялся. Начальник спустился вниз, нашел раствор хлорида палладия, который остался от прошлой корректировки, и добавил его весь. Потом посчитали, что это +15 мг/л. - Линия заработала. Медь пока тянется. Сейчас пишу и думаю, когда опять престанет работать: сегодня или через неделю. - Добавка паллалия ведь была минимальной. |

Внизу три колбы, в которые я по ошибке добавил концентрированную соляную кислоту |

Методика анализа ванны палладиевого активатора |
|
Методика определения олова сравнительно простая, но капризная. Йод окисляет двухвалентное олово до четырехвалентного, превращаясь при этом в бесцветный йодид-ион. Титрование ведут в среде соляной кислоты. Йодид в сильнокислой среде легко окисляется кислородом воздуха, что искажает результаты анализа. Чтобы изолировать раствор от воздуха, титрование ведут в атмосфере углекислого газа. Разумеется, никто баллон с СО2 ставить в заводской лаборатории не будет. Из положения выходят просто: в колбу добавляют немного соды или чистого мела (хуже). Реагируя с избытком кислоты, карбонаты дают углекислый газ, который вытесняет из раствора и колбы кислород. Главное не переборщить с количеством соды, иначе раствор вспенится и выплеснется из колбы. Потом проводят титрование.
К анализируемому раствору прикапывают титрант - 0.1N раствор йода. И ванна активации, и йод, имеет коричневый цвет, только раствор йода окрашен более интенсивно (плюс - йод имеет желтый оттенок, ванна - нет). Йод окисляет двухвалентное олово до четырехвалентного и обесцвечивается. От попадания очередной порции йода коричневый раствор в колбе на мгновение становится "более коричневым", но при перемешивании снова бледнеет (т.е. становится "менее коричневым"). По мере приближения точки эквивалентности при добавлении титранта раствор "коричневеет" все активнее, а при перемешивании бледнеет все медленнее. Коричневый анализируемый раствор и коричневый титрант. Как определить точку эквивалентности? В конце титрования в колбу добавляют раствор крахмала - с йодом он дает темно-синюю окраску. На практике окраска часто бывает не синей, а бурой, поскольку крахмал быстро гидролизует в сильнокислой среде до декстринов и дальше до глюкозы. Но переход окраски вполне различим (и с синим, и с бурым раствором) - когда от лишней капли йода раствор окрашивается в интенсивный цвет, который уже не исчезает при перемешивании. Кстати, не знаю почему, но после добавления соды в колбу с разведенной аликвотой ванны и кислотой, раствор немного бледнеет - видимо, частицы палладия частично оседают. Это также облегчает наблюдение за переходом окраски при последующем титровании. Когда сделал анализ ванны палладиевого активатора в первый раз, получилось, что олова меньше нормы. Где я допустил ошибку? Но оказалось, так и есть - в порядке эксперимента в ванне поддерживают заниженное содержание олова, - как и с палладием (см. предыдущую часть). Принцип: на чем еще можно сэкономить? Зачем мелочиться? - Сразу залили бы в ванну дистиллированную воду. Экономия будет максимальной. Хотя нет: можно залить воду из-под крана - так еще дешевле... |

Йодометрическое определение олова |

|

На этой фотографии показана немного другая ванна - "олово-висмут", но процедура аналогичная - добавление соды перед титрованием олова |

Конечная точка титрования |

|

|

|
|
Анализ соляной кислоты - простое кислотно-основное титрование от бесцветной до малиновой окраски фенолфталеина. Кстати, чем хороша соляная кислота - ее концентрация по мере работы ванны изменяется мало, два других компонента - палладий и олово постепенно расходуются.
|

Титрование соляной кислоты гидроксидом натрия |

|