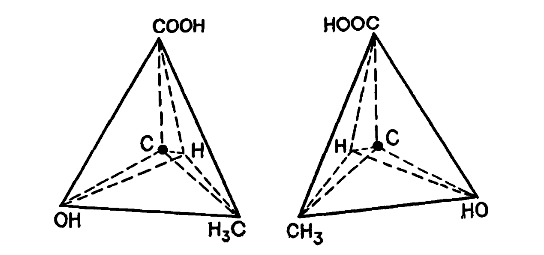
Теория валентности сыграла важнейшую роль в развитии теории химии вообще и органической химии в особенности. Исходя из теории валентности, Кекуле предположил, что атом углерода четырехвалентен, и в 1858 г. попытался, опираясь на это предположение, представить строение наиболее простых органических молекул и радикалов [5]. В том же 1858 г. шотландский химик Арчибальд Скотт Купер (1831-1892) предложил изображать силы, соединяющие атомы (или связи, как их принято называть), в виде черточек. После того как была «построена» первая органическая молекула, стало совершенно ясно, почему органические молекулы, как правило, значительно больше и сложнее, чем неорганические.
Согласно представлениям Кекуле, углеродные атомы могут соединяться друг с другом с помощью одной или нескольких из четырех своих валентных связей, образуя длинные цепи - прямые или разветвленные. По-видимому, никакие другие атомы не обладают этой замечательной способностью в той мере, в какой обладает ею углерод.
Итак, представив себе, что у каждого атома углерода четыре валентные связи, а у каждого атома водорода одна такая связь, можно изобразить три простейших углеводорода (соединения, молекулы которых образованы только атомами углерода и водорода), метан CH4, этан C2H6 и пропан C3H8, следующим образом:

Увеличивая число атомов углерода, эту последовательность можно продолжить, причем практически бесконечно. Добавляя к углеводородной цепи кислород (две валентные связи) или азот (три валентные связи), можно представить структурные формулы молекул этилового спирта (C2H6O) и метиламина (CH5N):
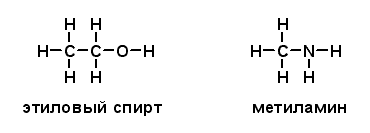
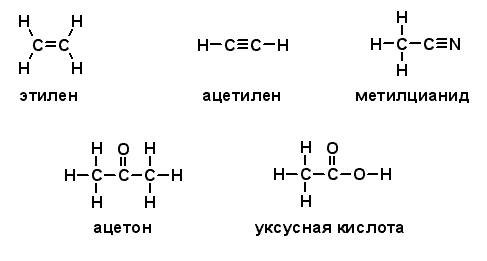
Допустив возможность наличия между соседними атомами двух связей (двойная связь) или трех связей (тройная связь), можно изобразить структурные формулы таких соединений, как этилен (C2H4), ацетилен (C2H2), метилцианид (C2H3N), ацетон (C3H6O) и уксусная кислота (C2H4O2):
Полезность структурных формул была настолько очевидной, что многие химики-органики приняли их сразу. Они признали полностью устаревшими все попытки изображать органические молекулы как структуры, построенные из радикалов. В результате было признано необходимым, записывая формулу соединения, показывать его атомную структуру.
Русский химик Александр Михайлович Бутлеров (1823-1886) использовал эту новую систему структурных формул в разработанной им теории строения органических соединений [6]. В 60-х годах прошлого столетия он показал, как с помощью структурных формул можно наглядно объяснить причины существования изомеров. Так, например, у этилового спирта и диметилового эфира одна и та же эмпирическая формула C2H6O, однако структурные формулы этих соединений значительно различаются:

поэтому не удивительно, что изменение в расположении атомов приводит к двум наборам очень разных свойств. В этиловом спирте один из шести атомов водорода присоединен к атому кислорода, в то время как в диметиловом эфире все шесть атомов водорода присоединены к атомам углерода. Атом кислорода удерживает атом водорода слабее, чем атом углерода, так что металлический натрий, добавленный к этиловому спирту, замещает водород (одну шестую общего количества). Натрий, добавленный к диметиловому эфиру, совсем не вытесняет водород. Таким образом, при составлении структурных формул можно руководствоваться химическими реакциями, а структурные формулы, в свою очередь, могут помочь понять суть реакций.
Бутлеров особенно много внимания уделил одному из типов изомерии, называемому таутомерией (динамической изомерией), при которой некоторые вещества всегда выступают как смеси двух соединений. Если одно из этих соединений выделить в чистом виде, оно сразу же частично перейдет в другое соединение. Бутлеров показал, что таутомерия обусловлена спонтанным переходом атома водорода от атома кислорода к соседнему атому углерода (и обратно).
Чтобы вполне доказать справедливость системы структурных формул, необходимо было определить структурную формулу бензола - углеводорода, содержащего шесть атомов углерода и шесть атомов водорода. Сделать это удалось далеко не сразу. Казалось, не существует такой структурной формулы, которая бы, отвечая требованиям валентности, в то же время объясняла бы большую устойчивость соединения. Первые варианты структурных формул бензола очень походили на формулы некоторых углеводородов - соединений весьма нестойких и не похожих по химическим свойствам на бензол.
Решить эту задачу смог опять-таки Кекуле. В один из дней 1865 г. (как он сам рассказывает) Кекуле в полудреме ехал в омнибусе, и ему пригрезилось, что он видит атомы, кружащиеся в танце. Вдруг конец одной цепи соединился с ее началом, и образовалось вращающееся кольцо. И Кекуле решил, что именно такой должна быть структурная формула бензола. До тех пор структурные формулы строились только в виде линейных цепей углеродных атомов, но теперь Кекуле ввел понятие «кольцо» (или «ядро») атомов углерода и предложил следующую структурную формулу бензола:

Это объяснение было принято, и представление о структурных формулах расширилось [7].
Согласно представлениям Кекуле, углеродные атомы могут соединяться друг с другом с помощью одной или нескольких из четырех своих валентных связей, образуя длинные цепи - прямые или разветвленные. По-видимому, никакие другие атомы не обладают этой замечательной способностью в той мере, в какой обладает ею углерод.
Итак, представив себе, что у каждого атома углерода четыре валентные связи, а у каждого атома водорода одна такая связь, можно изобразить три простейших углеводорода (соединения, молекулы которых образованы только атомами углерода и водорода), метан CH4, этан C2H6 и пропан C3H8, следующим образом:
Увеличивая число атомов углерода, эту последовательность можно продолжить, причем практически бесконечно. Добавляя к углеводородной цепи кислород (две валентные связи) или азот (три валентные связи), можно представить структурные формулы молекул этилового спирта (C2H6O) и метиламина (CH5N):
Допустив возможность наличия между соседними атомами двух связей (двойная связь) или трех связей (тройная связь), можно изобразить структурные формулы таких соединений, как этилен (C2H4), ацетилен (C2H2), метилцианид (C2H3N), ацетон (C3H6O) и уксусная кислота (C2H4O2):
Полезность структурных формул была настолько очевидной, что многие химики-органики приняли их сразу. Они признали полностью устаревшими все попытки изображать органические молекулы как структуры, построенные из радикалов. В результате было признано необходимым, записывая формулу соединения, показывать его атомную структуру.
Русский химик Александр Михайлович Бутлеров (1823-1886) использовал эту новую систему структурных формул в разработанной им теории строения органических соединений [6]. В 60-х годах прошлого столетия он показал, как с помощью структурных формул можно наглядно объяснить причины существования изомеров. Так, например, у этилового спирта и диметилового эфира одна и та же эмпирическая формула C2H6O, однако структурные формулы этих соединений значительно различаются:
поэтому не удивительно, что изменение в расположении атомов приводит к двум наборам очень разных свойств. В этиловом спирте один из шести атомов водорода присоединен к атому кислорода, в то время как в диметиловом эфире все шесть атомов водорода присоединены к атомам углерода. Атом кислорода удерживает атом водорода слабее, чем атом углерода, так что металлический натрий, добавленный к этиловому спирту, замещает водород (одну шестую общего количества). Натрий, добавленный к диметиловому эфиру, совсем не вытесняет водород. Таким образом, при составлении структурных формул можно руководствоваться химическими реакциями, а структурные формулы, в свою очередь, могут помочь понять суть реакций.
Бутлеров особенно много внимания уделил одному из типов изомерии, называемому таутомерией (динамической изомерией), при которой некоторые вещества всегда выступают как смеси двух соединений. Если одно из этих соединений выделить в чистом виде, оно сразу же частично перейдет в другое соединение. Бутлеров показал, что таутомерия обусловлена спонтанным переходом атома водорода от атома кислорода к соседнему атому углерода (и обратно).
Чтобы вполне доказать справедливость системы структурных формул, необходимо было определить структурную формулу бензола - углеводорода, содержащего шесть атомов углерода и шесть атомов водорода. Сделать это удалось далеко не сразу. Казалось, не существует такой структурной формулы, которая бы, отвечая требованиям валентности, в то же время объясняла бы большую устойчивость соединения. Первые варианты структурных формул бензола очень походили на формулы некоторых углеводородов - соединений весьма нестойких и не похожих по химическим свойствам на бензол.
Решить эту задачу смог опять-таки Кекуле. В один из дней 1865 г. (как он сам рассказывает) Кекуле в полудреме ехал в омнибусе, и ему пригрезилось, что он видит атомы, кружащиеся в танце. Вдруг конец одной цепи соединился с ее началом, и образовалось вращающееся кольцо. И Кекуле решил, что именно такой должна быть структурная формула бензола. До тех пор структурные формулы строились только в виде линейных цепей углеродных атомов, но теперь Кекуле ввел понятие «кольцо» (или «ядро») атомов углерода и предложил следующую структурную формулу бензола:
Это объяснение было принято, и представление о структурных формулах расширилось [7].