Обнаружив ошибку на странице, выделите ее и нажмите Ctrl + Enter
Глава 9. Физическая химия
В XVII и XVIII вв. мир химии и мир физики разделяла четкая граница. Химия изучала процессы, сопровождающиеся изменением молекулярной структуры, в то время как физика изучала такие процессы, которые подобными изменениями не сопровождались.
В начале XIX столетия, когда Дэви разрабатывал классификацию молекул неорганических соединений, а Бертло - классификацию молекул органических соединений, физики изучали потоки теплоты, другими словами - термодинамику (от греческого - движение тепла).
Выдающихся успехов в этой области достигли английский физик Джеймс Прескотт Джоуль (1818-1889) и немецкие физики Юлиус Роберт Майер (1814-1878) и Герман Людвиг Фердинанд Гельмгольц (1821-1894). К 40-м годам прошлого столетия в результате проведенных ими работ стало ясно, что в процессе перехода одной формы энергии в другую энергия не создается и не исчезает. Этот принцип получил название закона сохранения энергии, или первого начала термодинамики.
В своих работах французский физик Никола Леонар Сади Карно (1796-1832), английский физик Уильям Томсон, впоследствии лорд Кельвин (1824-1907), и немецкий физик Рудольф Джулиус Эмануэль Клаузиус (1822-1888) развили механическую теорию теплоты. Было показано, что при самопроизвольном переходе теплоты от точки с более высокой температурой к точке с более низкой температурой работа производится только в случае существенной разности температур, ибо часть теплоты неизбежно рассеивается в окружающую среду. Этот вывод можно обобщить и распространить на любой вид энергии.
В 1850 г. Клаузиус, пытаясь найти соотношение между количеством теплоты в изолированной системе и абсолютной температурой этой системы, ввел термин энтропия. Он показал, что при любых самопроизвольных изменениях энергии энтропия системы должна увеличиваться. Этот принцип был назван вторым началом термодинамики.
Естественно, что такого рода открытия не могли не повлиять на развитие химии. Ведь в конечном итоге основными источниками теплоты в XIX в. (кроме Солнца) были химические реакции: горение дерева, угля и нефти. Химикам было также известно, что теплота выделяется и при других химических реакциях, например при нейтрализации кислот основаниями, и что практически все химические реакции сопровождаются тем или иным тепловым эффектом: выделением теплоты (а иногда и света) или поглощением теплоты (а иногда и света).
В 1840 г. после опубликования работ русского химика Германа Ивановича Гесса (1802-1850) [1] граница между миром физики и химии была разрушена, и началось сотрудничество двух наук. Тщательно измерив действительное количество теплоты, выделяемой в процессе химических реакций между определенными количествами веществ, Гесс показал, что количество теплоты, получаемой (или поглощаемой) при переходе от одного вещества к другому, всегда одинаково и не зависит от того, с помощью какой химической реакции или сколькими этапами осуществляется этот переход. Благодаря этому обобщению (закон Гесса) Гесса иногда считают основателем термохимии (теплохимии).
Исходя из закона Гесса, представлялось вполне вероятным, что закон сохранения энергии равно применим и к химическим, и к физическим процессам. И действительно, дальнейшие обобщения показали, что законы термодинамики, вероятнее всего, проявляются в химии точно так же, как и в физике.
Это направление в экспериментах и в теории привело к выводу, что определенным химическим реакциям, как и физическим процессам, присуще свойственное только им самопроизвольное направление, приводящее к увеличению энтропии. Однако энтропия представляет собой величину, трудную для непосредственного измерения, поэтому химики начали искать другой, более простой критерий.
В 60-х годах прошлого столетия Бертло, уже завоевавший известность как органик-синтетик, обратился к термохимии. Он разработал методику проведения химических реакций в замкнутых сосудах, погруженных в воду заданной температуры. Определив температуру этой воды в конце реакции, можно было установить, какое количество теплоты выделяется в ходе данной реакции.
Используя такой калориметр (от латинского calorimeter - измерение тепла), Бертло тщательно измерил количество теплоты, выделяемой в результате сотен различных химических реакций. Подобные эксперименты независимо от Бертло провел также датский химик Ханс Петер Юрген Юлиус Томсен (1826-1909).
Бертло полагал, что реакции, сопровождающиеся выделением теплоты, являются самопроизвольными, в то время как реакции, сопровождающиеся поглощением теплоты, таковыми не являются. Поскольку каждая реакция, в ходе которой выделяется теплота, должна сопровождаться, если заставить ее идти в обратном направлении, поглощением теплоты (первыми стали придерживаться такой точки зрения Лавуазье и Лаплас), то, следовательно, любая химическая реакция идет самопроизвольно только в одном направлении, и при этом она сопровождается выделением теплоты.
Например, когда водород взаимодействует с кислородом, образуя воду, реакция протекает с выделением большого количества теплоты. Эта реакция самопроизвольная, и, однажды начавшись, она быстро идет к завершению и иногда заканчивается сильным взрывом.
В то же время обратная реакция - расщепление воды на водород и кислород - требует затраты энергии (тепловой или, лучше, электрической). Расщепление молекулы воды не является самопроизвольным; в отсутствие энергии расщепление вообще не происходит, и уже начавшаяся реакция тотчас же прекратится, если подачу энергии прервать.
Но это правило Бертло, на первый взгляд представлявшееся вполне приемлемым, было ошибочным. Во-первых, не все самопроизвольные реакции протекают с выделением теплоты; некоторые реакции сопровождаются поглощением теплоты, и в ходе таких реакций температура среды, окружающей реакционную смесь, действительно понижается.
Во-вторых, существуют обратимые реакции. Так, например, вещества A и B могут самопроизвольно взаимодействовать и превращаться в вещества C и D, которые в свою очередь могут вновь самопроизвольно образовать вещества A и B. И это несмотря на то, что если какая-либо реакция сопровождается выделением теплоты, то обратная ей реакция должна сопровождаться поглощением теплоты. Например, иодид водорода разлагается на йод и водород, которые вновь могут образовывать иодид водорода:
(две стрелки, направленные в противоположные стороны, показывают, что реакция обратима).
Во времена Бертло обратимые реакции были уже известны. В 1850 г. Уильямсон первым тщательно изучил их. Основываясь на результатах проведенных им работ, Уильямсон предложил структурные формулы эфиров. Он нашел условия, при которых смесь веществ A и B образовывала вещества C и D, а смесь веществ C и D образовывала вещества A и B. Однако и в том, и в другом случае в итоге получалась смесь веществ A, B, C и D, причем соотношение этих компонентов было определенным. Смесь при этом находилась в состоянии равновесия.
Хотя состав смеси оставался скорее всего постоянным, тем не менее Уильямсон считал, что вещества A и B реагируют, образуя вещества C и D, а вещества C и D реагируют, образуя вещества A и B. Обе реакции идут непрерывно, но они нейтрализуют друг друга, создавая иллюзию покоя, тогда как в действительности смесь находится в состоянии динамического равновесия.
Работа Уильямсона ознаменовала начало изучения химической кинетики - области химии, изучающей скорости химических реакций. Уильямсон ясно показал, что самопроизвольный характер химической реакции в ряде случаев определяет не просто выделение теплоты, а нечто большее. Проводя свои многочисленные калориметрические измерения, Бертло и Томсен уже выявили это «нечто большее», но, к сожалению, вопрос остался нерешенным из-за того, что работы Томсена были опубликованы на малодоступном ученым норвежском языке.
В начале XIX столетия, когда Дэви разрабатывал классификацию молекул неорганических соединений, а Бертло - классификацию молекул органических соединений, физики изучали потоки теплоты, другими словами - термодинамику (от греческого - движение тепла).
Выдающихся успехов в этой области достигли английский физик Джеймс Прескотт Джоуль (1818-1889) и немецкие физики Юлиус Роберт Майер (1814-1878) и Герман Людвиг Фердинанд Гельмгольц (1821-1894). К 40-м годам прошлого столетия в результате проведенных ими работ стало ясно, что в процессе перехода одной формы энергии в другую энергия не создается и не исчезает. Этот принцип получил название закона сохранения энергии, или первого начала термодинамики.
В своих работах французский физик Никола Леонар Сади Карно (1796-1832), английский физик Уильям Томсон, впоследствии лорд Кельвин (1824-1907), и немецкий физик Рудольф Джулиус Эмануэль Клаузиус (1822-1888) развили механическую теорию теплоты. Было показано, что при самопроизвольном переходе теплоты от точки с более высокой температурой к точке с более низкой температурой работа производится только в случае существенной разности температур, ибо часть теплоты неизбежно рассеивается в окружающую среду. Этот вывод можно обобщить и распространить на любой вид энергии.
В 1850 г. Клаузиус, пытаясь найти соотношение между количеством теплоты в изолированной системе и абсолютной температурой этой системы, ввел термин энтропия. Он показал, что при любых самопроизвольных изменениях энергии энтропия системы должна увеличиваться. Этот принцип был назван вторым началом термодинамики.
Естественно, что такого рода открытия не могли не повлиять на развитие химии. Ведь в конечном итоге основными источниками теплоты в XIX в. (кроме Солнца) были химические реакции: горение дерева, угля и нефти. Химикам было также известно, что теплота выделяется и при других химических реакциях, например при нейтрализации кислот основаниями, и что практически все химические реакции сопровождаются тем или иным тепловым эффектом: выделением теплоты (а иногда и света) или поглощением теплоты (а иногда и света).
В 1840 г. после опубликования работ русского химика Германа Ивановича Гесса (1802-1850) [1] граница между миром физики и химии была разрушена, и началось сотрудничество двух наук. Тщательно измерив действительное количество теплоты, выделяемой в процессе химических реакций между определенными количествами веществ, Гесс показал, что количество теплоты, получаемой (или поглощаемой) при переходе от одного вещества к другому, всегда одинаково и не зависит от того, с помощью какой химической реакции или сколькими этапами осуществляется этот переход. Благодаря этому обобщению (закон Гесса) Гесса иногда считают основателем термохимии (теплохимии).
Исходя из закона Гесса, представлялось вполне вероятным, что закон сохранения энергии равно применим и к химическим, и к физическим процессам. И действительно, дальнейшие обобщения показали, что законы термодинамики, вероятнее всего, проявляются в химии точно так же, как и в физике.
Это направление в экспериментах и в теории привело к выводу, что определенным химическим реакциям, как и физическим процессам, присуще свойственное только им самопроизвольное направление, приводящее к увеличению энтропии. Однако энтропия представляет собой величину, трудную для непосредственного измерения, поэтому химики начали искать другой, более простой критерий.
В 60-х годах прошлого столетия Бертло, уже завоевавший известность как органик-синтетик, обратился к термохимии. Он разработал методику проведения химических реакций в замкнутых сосудах, погруженных в воду заданной температуры. Определив температуру этой воды в конце реакции, можно было установить, какое количество теплоты выделяется в ходе данной реакции.
Используя такой калориметр (от латинского calorimeter - измерение тепла), Бертло тщательно измерил количество теплоты, выделяемой в результате сотен различных химических реакций. Подобные эксперименты независимо от Бертло провел также датский химик Ханс Петер Юрген Юлиус Томсен (1826-1909).
Бертло полагал, что реакции, сопровождающиеся выделением теплоты, являются самопроизвольными, в то время как реакции, сопровождающиеся поглощением теплоты, таковыми не являются. Поскольку каждая реакция, в ходе которой выделяется теплота, должна сопровождаться, если заставить ее идти в обратном направлении, поглощением теплоты (первыми стали придерживаться такой точки зрения Лавуазье и Лаплас), то, следовательно, любая химическая реакция идет самопроизвольно только в одном направлении, и при этом она сопровождается выделением теплоты.
Например, когда водород взаимодействует с кислородом, образуя воду, реакция протекает с выделением большого количества теплоты. Эта реакция самопроизвольная, и, однажды начавшись, она быстро идет к завершению и иногда заканчивается сильным взрывом.
В то же время обратная реакция - расщепление воды на водород и кислород - требует затраты энергии (тепловой или, лучше, электрической). Расщепление молекулы воды не является самопроизвольным; в отсутствие энергии расщепление вообще не происходит, и уже начавшаяся реакция тотчас же прекратится, если подачу энергии прервать.
Но это правило Бертло, на первый взгляд представлявшееся вполне приемлемым, было ошибочным. Во-первых, не все самопроизвольные реакции протекают с выделением теплоты; некоторые реакции сопровождаются поглощением теплоты, и в ходе таких реакций температура среды, окружающей реакционную смесь, действительно понижается.
Во-вторых, существуют обратимые реакции. Так, например, вещества A и B могут самопроизвольно взаимодействовать и превращаться в вещества C и D, которые в свою очередь могут вновь самопроизвольно образовать вещества A и B. И это несмотря на то, что если какая-либо реакция сопровождается выделением теплоты, то обратная ей реакция должна сопровождаться поглощением теплоты. Например, иодид водорода разлагается на йод и водород, которые вновь могут образовывать иодид водорода:
H2 + I2 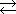 2HI
2HI
(две стрелки, направленные в противоположные стороны, показывают, что реакция обратима).
Во времена Бертло обратимые реакции были уже известны. В 1850 г. Уильямсон первым тщательно изучил их. Основываясь на результатах проведенных им работ, Уильямсон предложил структурные формулы эфиров. Он нашел условия, при которых смесь веществ A и B образовывала вещества C и D, а смесь веществ C и D образовывала вещества A и B. Однако и в том, и в другом случае в итоге получалась смесь веществ A, B, C и D, причем соотношение этих компонентов было определенным. Смесь при этом находилась в состоянии равновесия.
Хотя состав смеси оставался скорее всего постоянным, тем не менее Уильямсон считал, что вещества A и B реагируют, образуя вещества C и D, а вещества C и D реагируют, образуя вещества A и B. Обе реакции идут непрерывно, но они нейтрализуют друг друга, создавая иллюзию покоя, тогда как в действительности смесь находится в состоянии динамического равновесия.
Работа Уильямсона ознаменовала начало изучения химической кинетики - области химии, изучающей скорости химических реакций. Уильямсон ясно показал, что самопроизвольный характер химической реакции в ряде случаев определяет не просто выделение теплоты, а нечто большее. Проводя свои многочисленные калориметрические измерения, Бертло и Томсен уже выявили это «нечто большее», но, к сожалению, вопрос остался нерешенным из-за того, что работы Томсена были опубликованы на малодоступном ученым норвежском языке.